Proprietary Technology
Innovative Proprietary Technology
The Dual-TCE (Thermo-Chemical Electrolysis)
Hydrogen is the best energy carrier for a carbon-free world. There is no CO2 produced when hydrogen is burned. However, a significant amount of hydrogen is produced today using fossil-fuel-intensive technologies that emit massive volumes of CO2, undermining the promise of global decarbonization.
Hydrogen can also be extracted from water. Electrolysis is the name for this procedure, which necessitates the use of electricity. If the electricity comes from a renewable source, this procedure produces no CO2. Unfortunately, water electrolysis is inefficient and expensive, making it uncompetitive with traditional fossil fuel extraction methods. Our goal is to develop a water-splitting technology that can compete with fossil fuels in terms of cost.

Energy Storage
To complete the whole transition to renewable energy, we must store energy when we have it and use it when we need it. For instance, solar energy can be stored during the day and used at night, or even from summer to winter.
Solar and wind energy can be stored in hydrogen, which can then be converted into electricity when necessary. Batteries, for example, are another technique to store energy. Growing storage capacity with batteries, on the other hand, necessitates the purchase of new batteries, whereas increasing storage capacity with hydrogen necessitates the purchase of additional tanks.
Challenges of Electrolysis
Water electrolysis is not a new concept. It was first discovered in 1789 and has been employed in commercial applications for more than a century. Despite the advancements made in upgrading this centuries-old technology throughout the years, it still has fundamental flaws that limit widespread adoption:
High Equipment Cost
Membranes contribute to the complexity of electrolyzers, resulting in higher production costs. PEM (Polymer Electrolyte Membrane) electrolyzers necessitate the use of precious metals, which raises the price. External compressors are needed because of the low hydrogen pressure, and cooling is needed because of the heat created by the inefficient process, which increases the equipment cost.
Low Efficiency
The efficiency of modern electrolysis is 60-70 %. The cost of green hydrogen rises rapidly as a result of this low efficiency.
Safety
Electrolysis produces both hydrogen and oxygen at the same time, potentially resulting in a dangerous and explosive mixture.
Dual-TCE (Thermo-Chemical Electrolysis) is a ground-breaking technology for water splitting. Electrolytic hydrogen production has technological hurdles in terms of increasing efficiency, economic value, and global integration potential. The water oxidation and reduction reactions in traditional water electrolysis are time and space linked because they occur simultaneously at an anode and a cathode in the same cell.
There are challenges, such as product dispersion, and this is placing important limitations on material selection and processing conditions. We decouple the specific reactions by splitting the process into two stages: an electrochemical stage that reduces water at the cathode and oxidizes the anode, and a spontaneous chemical stage that reduces the anode back to its initial state by oxidizing water at a higher temperature. In a membrane-free, two-electrode cell, this enables total water splitting. This will help us to generate hydrogen in a simple, cyclic process that is high in efficiency, durability, safety, and scale-up potential.
Dual-TCE is a novel method of creating hydrogen from water. Climate change is the most pressing issue confronting our age. We must reduce CO2 emissions from fossil fuels to net zero as soon as possible, or we will experience serious consequences. Green hydrogen, an ideal fossil fuel alternative, is the key to achieving these global goals. You can burn it like fossil fuel or use a fuel cell to convert it into energy with no CO2 emissions, simply water.
When compared to the 75 % efficiency of a standard water electrolysis process, Dual-TCE is the first technology to provide energy efficiency of 95 %. Dual-TCE devices are less expensive, more scalable, safer, and have a higher operating pressure. Based on the technology development, we believe that in the near future, we will be able to heat our homes, power automobiles, ships, planes, and fuel industries using green and environmentally beneficial hydrogen in a cost-efficient manner.
The Dual-TCE system's electrochemical process is based on a characteristic of nickel hydroxide (Ni (OH)2), a substance commonly utilized in rechargeable electric batteries. When hydroxide ions arrive at an anode made of Ni (OH)2, a chemical reaction occurs in which the Ni (OH)2 is transformed into nickel oxyhydroxide (NiOOH) plus a number of free electrons that can move to the cathode. This process proceeds until the quantity of Ni (OH)2 molecules that can be transformed to NiOOH becomes scarce. The circuit in the Dual-TCE system is disrupted and the electric current stops flowing when that point is reached. The NiOOH molecules spontaneously break down into Ni (OH)2 and O2 in that environment.
Dual-TCE uses electricity to split water into hydrogen and oxygen, similar to electrolysis. Unlike traditional electrolysis, however, hydrogen and oxygen are produced separately in two stages: electrochemical and thermo-chemical electrolysis (TCE).
Membrane-free electrolytic reactors from Dual-TCE are well-suited to high-pressure hydrogen production and cost-effective scaling. This disruptive technique allows for the creation of green hydrogen while maintaining high energy efficiency inside the reactors and a 95 % system efficiency.
Why Dual-TCE?
Affordable: Dual-TCE separates the hydrogen and oxygen evolution reactions into two phases, guaranteeing that hydrogen and oxygen do not combine. This eliminates the need for the membrane, which is the most expensive and sensitive component of an electrolyzer. As a result, Dual-TCE devices are easier and less expensive to manufacture, lowering CAPEX significantly (Capital Expenditure). Dual-TCE enables high-pressure hydrogen generation (100+ bar), minimizing the requirement for compressors and lowering CAPEX and OPEX (Operating Expense) associated with compression.
Safe: Dual-TCE generates hydrogen and oxygen in two phases, avoiding contact between the two gases and thus avoiding the potential of explosive mixture.
Efficient: Both hydrogen and oxygen are generated when water is split. Electricity-based oxygen generation (electrochemical) is intrinsically inefficient, resulting in a 25% power loss. Due to the fact that Dual-TCE creates oxygen thermally, there is essentially little power loss. The outcome is high energy efficiency.
High Pressure and Scale-Up
The lack of a membrane leads to high hydrogen generation of up to 100 bars (without a compressor). Furthermore, because the hydrogen generation electrochemical process and the oxygen generation chemical process are separated, the partial-load operation is possible without the risk of H2/O2 mixing, making Dual-TCE water splitting more compatible with renewable energy sources like solar and wind than traditional water electrolysis.
Green hydrogen production at high pressure (ideal for industrial applications) and high efficiency is possible with the Dual-TCE technology. Additionally, because the Dual-TCE water splitting cells lack membrane separators, the technique is very straightforward to scale up and requires less maintenance than traditional water electrolysis, resulting in much lower CAPEX and OPEX expenses.
Electrochemical – Chemical Water Splitting
The method we developed is represented in Fig. 1, which contrasts alkaline water electrolysis (Fig. 1a) with our suggested technique (Fig. 1b). Because the fuel production reaction (hydrogen evolution in the case of water splitting) occurs at the cathode, our technique basically alters the water oxidation reaction at the anode. As a result, at the cathode, water is converted to hydrogen gas, releasing OH– ions.
In a thermo-chemical electrolysis stage, the four-electron oxygen evolution reaction (OER), which occurs at the anode in conventional electrolysis (Fig. 1a), is divided into two steps: four one-electron oxidation reactions of nickel hydroxide (Ni (OH)2) anode, followed by spontaneous oxygen evolution and anode regeneration.
The O-O bond is formed during the second stage of our dual-stage electrochemical–thermally chemical electrolysis (Dual-TCE) cycle, when the oxidized anode (already nickel oxyhydroxide (NiOOH)) is reduced to Ni (OH)2 in a spontaneous, exothermic chemical reaction that produces O2 and regenerates the anode, completing the overall water-splitting reaction. Our strategy uses four metal centers to store oxidative equivalents, similar to the oxygen-evolving complex in natural systems, which serves to level the potential of the elementary oxidation processes, minimizing the overall reaction over potential. Our approach, as compared to earlier work in this developing industry, enables decoupled water splitting with increased efficiency and versatility.
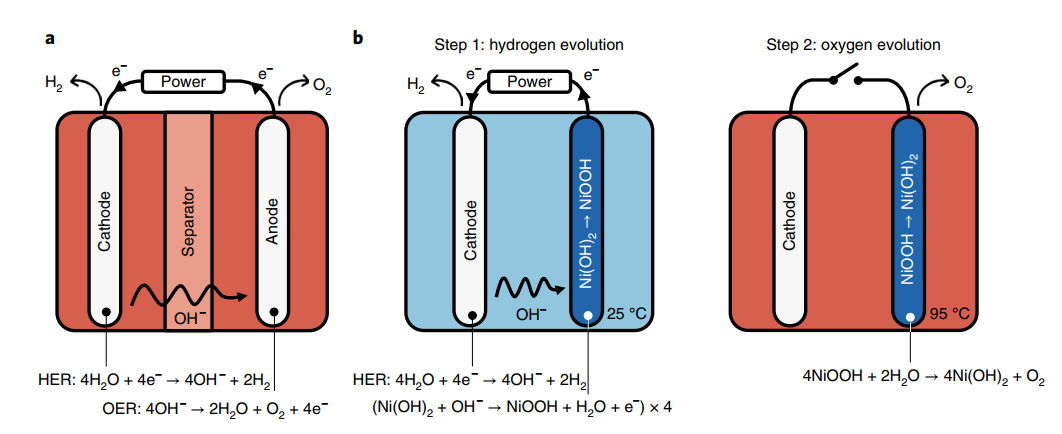
Fig. 1 | Schematic of alkaline water electrolysis and the Dual-TCE water-splitting process. a, In alkaline water electrolysis, which typically takes place at elevated temperatures (50–80 °C)6, the OER and HER are coupled in both time and space, as they occur simultaneously at an anode and a cathode, which are placed together in the same cell. A diaphragm or anion exchange membrane separates the anode and cathode compartments and prevents O2/H2 crossover. b, Dual-TCE water splitting proceeds in two consecutive steps. An electrochemical step (left) reduces water by the conventional HER at the cathode, liberating hydroxide ions (OH–) that oxidize a nickel hydroxide (Ni(OH)2) anode into nickel oxyhydroxide (NiOOH). This step is followed by a chemical step (right), wherein the NiOOH anode reacts with water to spontaneously produce oxygen. The first (electrochemical) reaction occurs at ambient temperature (~25 °C), whereas the second (chemical) reaction proceeds at elevated temperatures (~95 °C) for the optimum rate of reaction. The first and second reactions sum up to the overall water-splitting reaction, 2H2O → 2H2 + O2.
Connecting Dual-TCE production process with renewable energy systems
The electrolysis process uses electricity to split water into hydrogen and oxygen. When renewable power is used for this process, hydrogen becomes a complementary carrier of renewable energy.
Hydrogen produced with excess solar PV and wind power can be stored for later use – as a fuel for transport, industry and other sectors. Hydrogen production can be used as a ‘smart’ load to increase power system flexibility and help to decarbonise the overall economy.
Our aim also is to have hydrogen production using renewable electricity and to reach 12 EJ (exajoule) in 2035.
Hydrogen produced during the process of electrolysis can be used as a medium for energy storage and for applications such as producing heat for buildings, refueling fuel cell vehicles and as a source of feedstock for industry.
Producing hydrogen from renewable energy helps avoid slowdown and hence increases the return on investment for variable renewable energy (VRE) asset owners, especially when there is extra renewable electricity generation in the system.
When renewable energy cannot be fed into the electricity grid owing to network problems or low demand, it can be fed to electrolyzers for electrolysis to produce hydrogen. Excess renewable energy (which is likely to be curtailed or sold at near-zero marginal pricing) can greatly enhance hydrogen production economics. Excess renewable energy is also sold, allowing VRE asset owners to earn additional cash and lessen their exposure to power price volatility.
The cost of electricity accounts for a significant portion of the total cost of creating hydrogen by electrolysis. The contribution of electricity to the overall cost of hydrogen is determined by the cost of power, the size of the installation, load hours, and the electrolyzer’s location. While the cost of electricity accounts for about 30% of the total cost of creating hydrogen, it can reach 60% in rare situations.
Electrolyzer load factors are directly proportional to the levelized cost of hydrogen (LCOH) (USD/ KgH2). The lower the share of fixed cost and the larger the share of electricity cost in the LCOH, the higher the load factor. As a result, decreased electricity costs, such as those resulting from the use of excess renewable energy, improve the cost-effectiveness of electrolytic hydrogen. For this reason alone, we are merging renewable energy with the Dual-TCE system to gain a competitive advantage.
Potential Economic Impact
Our team compares the Dual TCE's 95 % energetic efficiency to a current-methods baseline of 75 %. A 20-percentage-point increase in energy efficiency sounds significant, and it is. It means that with the same inputs, such as the same physical equipment and the same quantity of electricity provided by solar panels or wind turbines, a given installation will produce 25% more hydrogen.
Another important benefit of the Dual-TCE system, according to our experts, is its membrane-free architecture. Due to the lack of a membrane or diaphragm, the capital cost of the equipment should be reduced by as much as 50% compared to present electrolyzers. Dual-TCE electrolyzers will also be able to operate at higher pressures than present electrolyzers, allowing for easier integration with downstream processes.
Electrode Design
In a phrase, our Dual-TCE water-splitting method substitutes the traditional alkaline electrolysis water oxidation reaction with a two-stage cycle in which the anode is first charged (electrochemically) and then regenerated (chemically). The hydrogen evolution reaction (HER) in the first step of our technique is identical to that of alkaline electrolysis, with the exception that it takes place under moderate temperature rather than at elevated temperatures (usually 50–80 °C). As a result, in our approach, the same cathode materials that are used in alkaline electrolysis, such as Raney nickel or other HER catalysts, can be applied. For our proof-of-concept investigations, we employ platinized nickel-coated stainless-steel mesh cathodes as a standardized benchmark HER cathode.
The anode, which acts differently in our technique than in traditional alkaline electrolysis, necessitates careful material selection and optimization. Consistency in alkaline solutions; cyclability of the metal hydroxide and oxyhydroxide phases; a redox potential higher than the reversible OER potential (1.23 VRHE) but lower than the potential at which oxygen evolves on the anode material; high capacity; fast charging; and a fast rate of regeneration are some of the key selection criteria for the anode. All of these factors point to Ni (OH)2/ NiOOH anodes, which are widely employed in alkaline secondary batteries due to their high energy density and cyclability.
Despite the fact that the Ni (OH)2/NiOOH standard redox potential is 190 mV greater than the reversible OER potential, the OER is kinetically suppressed by its significant overpotential. As long as alkaline batteries are not overcharged, they can run hundreds of cycles without parasitic oxygen development, which causes swelling and malfunction. Additives that catalyze the Ni (OH)2/NiOOH reaction and limit oxygen evolution are utilized to decrease parasite oxygen evolution that can occur during overcharging. Cobalt is one of the most often utilized additives.
The anode's electron and proton conductivity strengthen with cobalt, and the Ni (OH)2/NiOOH redox potential moves cathodically, extending the potential window between charging and overcharging. As a result, Ni1-xCox (OH)2 anodes can be charged to a higher state of charge (SOC) at a lower voltage than their undoped Ni (OH)2 counterparts without emitting oxygen. Unlike battery anodes, which should avoid producing oxygen, our anodes must do so in the Dual-TCE cycle's second stage. During the hydrogen generation (first) stage, it is also necessary to restrict the spontaneous regeneration reaction, which is associated with oxygen evolution.
As a result, our anodes and operating conditions are designed to reduce oxygen evolution in the first stage while increasing oxygen generation in the second. Because ECI (electrochemical impregnation) electrodes have a larger surface area than sintered or pasted electrodes, they can handle higher current densities at lower overpotentials. Furthermore, when compared to pasted anodes, the ECI anodes have a substantially greater regeneration rate.
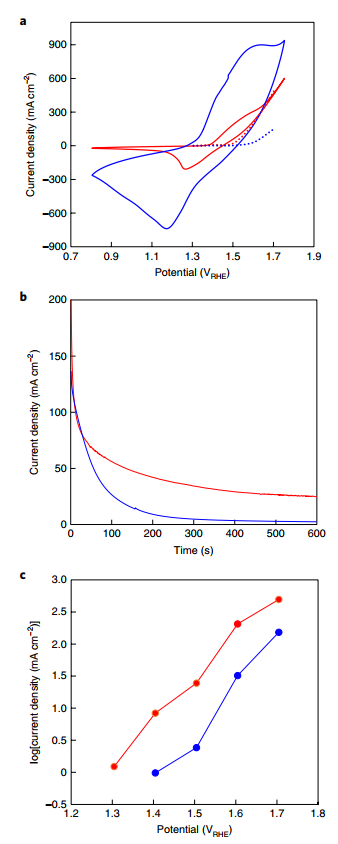
Fig. 2 | Electrochemical properties of Ni0.9Co0.1(OH)2 and NiFe LDH anodes. a, Cyclic voltammograms measured at a scan rate of 50 mV s-1. b, Chronoamperometric current traces at 1.48 VRHE. c, Tafel plots of the steady-state water oxidation current as a function of the potential, from 10-min amperograms, such as those presented in b. The dotted curves in a represent the steady-state water oxidation current extracted from c. For all graphs, the Ni0.9Co0.1(OH)2 anode is shown in blue and the NiFe LDH anode is shown in red. All tests were carried out in 5 M KOH at ambient temperature
Electrochemical Description
The open-circuit potential (OCP) of a Ni0.9Co0.1(OH)2 anode as a function of the SOC and the cyclic voltammogram were measured and compared to those of an undoped Ni (OH)2 anode. Over a SOC range of 10–90 percent, the OCP for the Ni0.9Co0.1(OH)2 anode is 1.31–1.41 V RHE, and for the undoped Ni (OH)2 anode is 1.37–1.42 VRHE. The inclusion of cobalt causes a 50-mV shift in the potential cathodically, reducing oxygen development at the anode during the hydrogen generating step.
The results were achieved using Ni 0.9Co0.1(OH)2 anodes. Our Ni0.9Co0.1(OH)2 anode's electrochemical characteristics were evaluated and compared to a benchmark water oxidation catalyst, NiFe layered double hydroxide (LDH). Figure 2a shows cyclic voltammograms of the Ni0.9Co0.1(OH)2 and NiFe LDH anodes. During the anodic sweep, the Ni0.9Co0.1(OH)2 anode exhibits a huge pseudo capacitive oxidation wave of Ni+2 to Ni+3, which begins at 1.3 VRHE and precedes the water oxidation currents of both anodes.
This demonstrates the ability to outperform the NiFe LDH water oxidation catalyst electrochemically. Chronogram perimetry measurements were performed at constant potentials between 1.3 and 1.7 V RHE to differentiate between the pseudo-capacitive anode oxidation current and the steady-state water oxidation current, as illustrated in Fig. 2b for 1.48 VRHE.
The Tafel plot of the steady-state water oxidation current of both anodes, shown in Fig. 2c and superimposed in Fig. 2a, is produced by the asymptotic values obtained at different potentials (dotted curves). The steady-state water oxidation current for the Ni0.9Co0.1(OH)2 anode is much smaller than the pseudo-capacitive current recorded in the cyclic voltammogram, showing that the pseudo-capacitive anode charging reaction is the dominating mechanism at these potentials. When comparing steady-state water oxidation and pseudo-capacitive currents for the NiFe LDH anode, water oxidation accounts for the majority of the net current.
Hydrogen Purity and Faradaic Efficiency
We compared the capacitive energy with the total charge that flows during chronoamperometric charging to calculate the Faradaic efficiency of the Ni0.9Co0.1(OH)2 anode charging reaction. The Faradaic efficiency was greater than 99.7% for more than 2,000s when charging a fully discharged anode at a potential of 1.48 VRHE, during which the anode was charged by more than 50 Ccm-2. The dissolved oxygen concentration in the electrolyte was monitored while charging a fully discharged anode at a constant voltage of 1.48 VRHE to ensure that no oxygen was generated.
Up to a charge of 55 Ccm-2, no increase in dissolved oxygen content was detected. The concentration of dissolved oxygen was also measured under conditions that were as close to the hydrogen generating phase of the Dual-TCE process as possible.
Under chronoamperometric charging of a regenerated Ni0.9Co0.1(OH)2 anode at 1.48 VRHE, the dissolved oxygen concentration was also determined. Up to 3Ccm-2, no increase in the dissolved oxygen content over the background level was recorded in either scenario, indicating that no oxygen was created while charging the anode.
Furthermore, on anode charging for 100 seconds at a constant current density of 50 mA cm-2, gas chromatography measurements revealed an increase in hydrogen concentration but no increase in oxygen concentration in the headspace of a sealed cell. These findings show that no parasitic side reactions, such as oxygen evolution, occur during the hydrogen generating step of the Dual-TCE process while charging the anode.
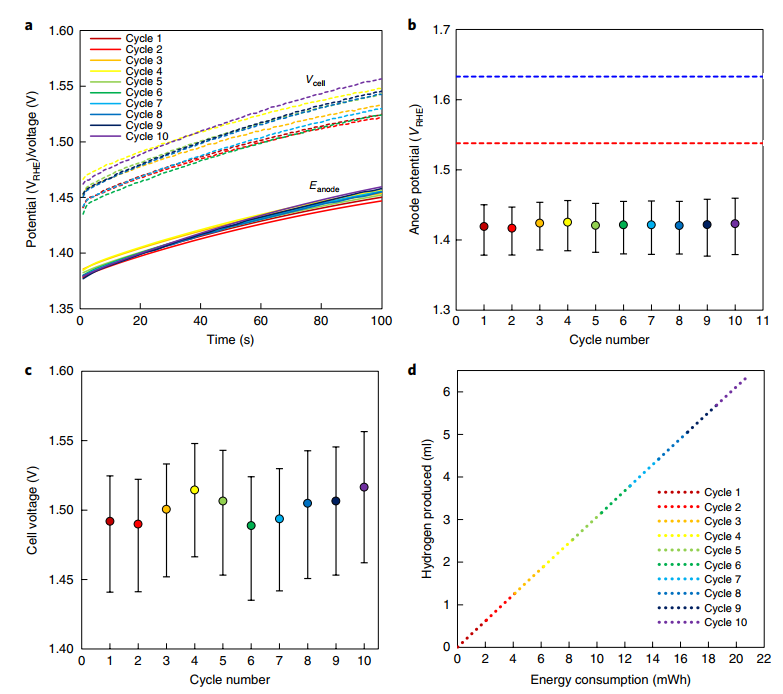
Fig. 3 | Dual-TCE water splitting in alkaline solution. a, Anode potential (Eanode, full lines) and cell voltage (Vcell, dashed lines) during the hydrogen generation steps of ten consecutive 100 s/100 s DUAL-TCE cycles at a nominal current density of 50 mA cm-2 in 5 M KOH solution. b, Corresponding average anode potential (Eanode, data points) and range (bars) in each cycle. c, Average cell voltage (Vcell, data points) and range (bars) in each cycle. d, Cumulative hydrogen production (at 1 atm and 25 °C) as a function of electrical energy consumption during the DUAL-TCE cycles. The blue and red dotted lines in b represent the potential of the OER reaction extracted from Fig. 2c at a steady-state current of 50 mA cm-2 of the Ni0.9Co0.1(OH)2 anode and NiFe LDH anode, respectively. The cycle-to-cycle variations in the cell voltage are attributed to the cycling procedure that involved disconnecting and reconnecting the anode manually between the steps, leading to small variations in the contact and series resistances.